Having produced and formed a metal, it is necessary to ensure that it performs well during service. A major consideration in this is corrosion. This involves loss of material from the metal’s surface and can be divided into two processes: dry oxidation and wet corrosion.
Oxidation and Corrosion of Metals
Dry Oxidation
The earth’s atmosphere is oxidising. Nearly all of the earth’s crust consists of oxides, which indicates that this is the preferred minimum energy state for most materials. Gold and silver are the only two metals that are found in their native, unoxidised state. The general oxidising reaction can be written as:
M + O → MO ……(1)
where M is the metal and O is oxygen.
For all metals, except gold and silver, this reaction is accompanied by release of energy, indicating the unstable nature of the metals. In fact this characteristic is shared with many other materials, which is why, for example, burning hydrocarbons is a useful source of heat. Normally the oxidation takes place in two steps.
First, the metal forms an ion, releasing electrons, and the electrons are accepted by oxygen to form an ion:
M → M2+ + 2e and O + 2e → O2− …….(2)
Secondly, the ions attract one another to form the oxide compound:
M2+ + O2− → MO ……..(3)
At the metal surface the oxygen ions attach themselves to the metal to form a thin layer of oxide. Thereafter, for the oxidation to continue, the metal M2+ ions and the electrons must diffuse outwards through this layer to form and meet more oxygen O2− ions at the outer surface, or the oxygen ions must diffuse inwards.
The rate of oxidation is determined by whichever reaction can proceed the faster and, largely, this is controlled by the thickness and structure of the oxide skin. On some metals the oxide occupies a lower volume than the metal from which it was formed. If it is brittle (and oxides usually are), it will crack and split, exposing fresh metal to more corrosion.
On other metals the oxide occupies a higher volume and it will tend to wrinkle and spring away, again exposing fresh metal. Even in these circumstances, the rates of reaction are generally low. In some other cases, however, the oxide volume matches the metal volume and thin adherent films form that act as near total barriers to further oxidation.
This is true of aluminium, which is why it does not need protection against corrosion when used, for example in window frames, and chromium and nickel, which are therefore the essential components of so-called ‘stainless steel’.
Wet Corrosion
In the presence of moisture the situation changes drastically and the loss of metal by corrosion becomes much more significant. Indeed, in several countries, including Japan, the UK and the USA, estimated losses to the national economy due to corrosion could be as high as up to 5% of their gross domestic product (GDP).
The explanation for the high corrosion rates is that the metal ions formed in equation (2) are soluble in the corroding medium (water); the electrons produced are then conducted through the metal to a nearby place where they are consumed in the reaction with oxygen and water to produce hydroxyl ions, which in turn link up with the metal ions to give a hydroxide.
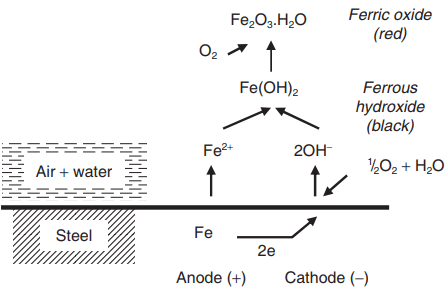
The corrosion of iron, illustrated in Fig. 1, is a useful example. The reaction in which the iron atoms pass into solution as Fe2+ ions leaving behind two electrons is called the anodic reaction and takes place at the anode of a resulting corrosion cell.
The hydroxyl ions are produced in the cathodic reaction, which takes place at the cathode of the cell, and the hydrated iron oxides (Fe(OH)2 or Fe(OH)3) are deposited either loosely on the metal surface or away from it, thus giving little or no protection. This, coupled with the ready conduction of the electrons through the iron, results in high rate of attack – many millions of times faster than that in air.
The Electromotive Series
Because wet oxidation involves electron flow in conductors, the application of an external voltage will either slow down or increase the rate of reaction depending on whether the applied voltage is negative or positive.
The values of the voltage that cause the anodic reaction to stop in different metals form the so-called electromotive series. By convention, the voltage is given in relation to that for the ionisation of hydrogen:
H → H+ + e …..(4)
This therefore appears as zero volts in the series. Some selected values from this are given in Table 1.
Those metals which are more positive than the reference value are anodic and will corrode, metals which are relatively cathodic will not. The series also tells us what happens when two different metals are connected while in an electrolyte. The more anodic metal will form the anode of the corrosion cell and hence will corrode, while the less anodic one forms the cathode and will not corrode. The resulting voltage difference between the metals can be useful, and was used in one of the first electrical batteries – the Daniell cell (Fig. 2), developed in 1836.
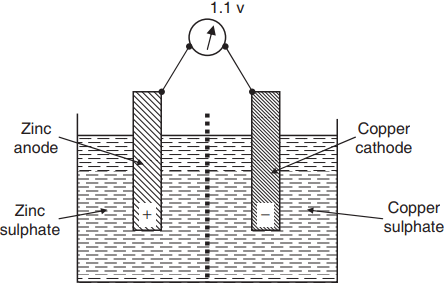
This consists of a zinc anode (which corrodes) in a zinc sulphate solution and a copper cathode (which does not corrode) in a copper sulphate solution; the two solutions are separated by a semi-permeable membrane, which prevents the copper ions in the copper sulphate solution from reaching the zinc anode and undergoing reduction. The voltage produced is the difference in standard electrode potential of the two elements i.e. 1.1v. The voltages in Table 1 must, however, be used with some caution.
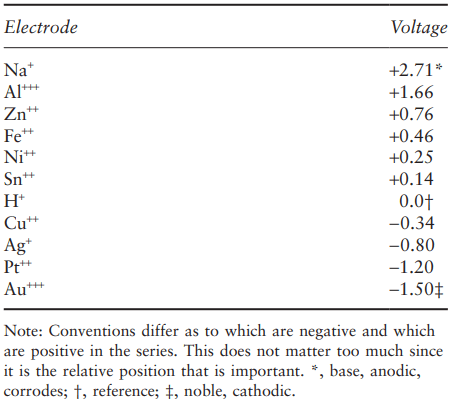
standard electrode potentials.
They apply in controlled laboratory conditions with the ions in solution having specific concentrations. In other environments e.g. seawater, and at other temperatures, the voltages will vary in magnitude, and even in order.
Despite this, they provide a very useful guide to performance. A further limitation of the electromotive series is that although it tells us about where corrosion is likely to occur, it tells us little about the likely corrosion rate.
For example, aluminium, which is high in the anodic voltage range, corrodes extremely slowly in moist atmospheres because a thin dry film of Al2O3 forms on the metal surface. However, in seawater the chloride ions tend to breakdown this protective film and aluminium corrodes very rapidly. Corrosion rates are also temperature dependent; for example mild steel in aerated water corrodes six times faster at 100°C than at 0°C.
Localised Corrosion
There are some circumstances in which wet corrosion can occur at selected localised sites. In these circumstances the corrosion can be intense and lead to premature failure of a component rather than generalised loss of material.
Intergranular attack: The grain boundaries in metals have different corrosion properties from the rest of the grain and can become the anodic region of the corrosion cell at which the corrosion is concentrated.
Concentration cell corrosion: The localised corrosion is a consequence of a difference in the constitution of the electrolyte itself. For example, consider water containing dissolved oxygen with differing concentrations in different regions. The reaction:
2H2O + O2 + 4e− → 4(OH)− …..(5)
removes electrons, and these must be supplied from adjacent areas, which then become deficient in oxygen. These act as the anode and hence corrode. Thus, in a bolted connection, corrosion will occur in the inaccessible (i.e. oxygen-poor) areas, e.g. under the bolt head (Fig. 3a).
A classic case is the ‘waterline’ corrosion of steel piling in stagnant water. Here the surface layers of the water are richer in oxygen and become the cathode. The lower, oxygen-deficient layers are anodic and corrosion occurs locally (Fig. 3b).
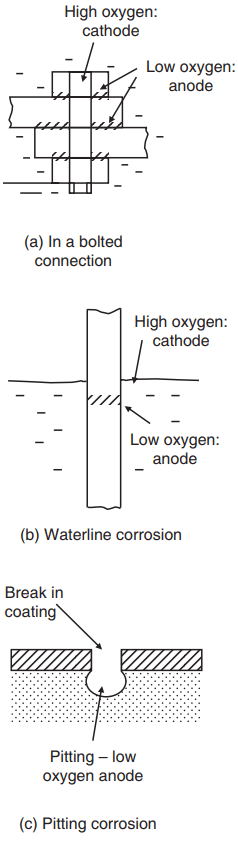
Much the same mechanism applies to pitting corrosion, which typically occurs where the metal is exposed at a break in a protective coating. The oxygen-poor region at the bottom of the pit is anodic and the pit therefore tends to deepen, often rapidly (Fig. 3c).
Note that waterline corrosion can be confused with a different phenomenon. We are all familiar with the enhanced corrosion that is seen on steel supports of seaside piers etc. Here we have a region that is washed by wave and tidal action. The region is alternately wetted and dried and it is this that accelerates corrosion.
Stress corrosion cracking: With some materials, when loaded or stressed in a corrosive environment, cracks can grow steadily under a stress intensity factor (K) that is much less than the critical stress intensity factor Kc.
This is clearly dangerous and can lead to brittle failure, even in a ductile material, after many years of apparently normal structural behaviour. Examples are stainless steels in chloride solutions and brass in ammonia.
Corrosion fatigue: The combination of cycling loading and a corrosive environment can lead to significant reductions in the fatigue life of a material, far greater than would be expected from the sum of the cyclic loading and the corrosion acting independently. With ferrous metals, the fatigue endurance limit also disappears, which makes safe design more difficult.
A classic situation is steel in sea water, which necessitates great care being taken when designing offshore structures, e.g. for oil and gas production, which will be subjected to wave action and seawater throughout their working life.
Thanks for reading about “oxidation and corrosion of metals”.