Degradation of Concrete
The degradation agencies that affect concrete can be divided into two broad groups:
- those whose action is initially chemical, before subsequently leading to loss of physical integrity; these include sulphates, seawater, acids and alkali– silica reactions
- those which directly lead to physical effects, such as freeze–thaw and fire.
We will now consider each of these in turn.
Attack by Sulphates
A controlled amount of calcium sulphate, in the form of gypsum, is added to Portland cement during its manufacture to control the setting process. Further sulphates in fresh concrete can arise from contaminated aggregates, a particular problem in some Middle Eastern countries.
Sources of sulphates that can penetrate hardened concrete include ground-water from some clay soils, fertilisers and industrial effluent, and so we can see that any problems mainly occur in concrete in contact with the ground e.g. in foundations, floor slabs and retaining walls. Sodium, potassium, calcium and magnesium sulphates are all common, and when in solution these will all react with components of the hardened cement paste.
The sulphates and the hydrated aluminate phases in the hardened cement paste react to form ettringite. Using the cement chemists’ shorthand notation, the reaction of calcium sulphate with the monosulphate hydrate is:
2CŠ + C3A.CŠ.12H + 20H → C3A.3CŠ.32H (1)
and with the direct hydrate:
3CŠ + C3A.6H + 26H → C3A.3CŠ.32H (2)
Both reactions are expansive, with the solid phases increasing significantly in volume, causing expansive forces and, possibly, disruption.
Sodium sulphate (NŠ.10H where N = Na2O) also forms ettringite by reacting with the hydrated aluminate:
3NŠ.10H + 2C3A.6H → C3A.3CŠ.32H + 2AH + 3NH + 5H (3)
but in addition it reacts with the calcium hydroxide in the HCP:
NŠ.10H + CH → CŠ.2H + NH + 8H
[NH = 2NaOH] (4)
This is analogous to acid attack, and in flowing water it is possible for the calcium hydroxide to be completely leached out. With magnesium sulphate (MŠ.7H where M = MgO), a similar reaction to (4) takes place, but the magnesium hydroxide formed is relatively insoluble and poorly alkaline; this reduces the stability of the calcium silicate hydrate, which is also attacked:
C3S2H3 + 3MŠ.7H → 3CŠ.2H + 3MH +2S.aq (5)
Since it is the calcium silicate hydrate that gives the hardened cement its strength, attack by magnesium sulphate can be more severe than that by other sulphates.
In each case, attack occurs only when the amount of sulphate present exceeds a certain threshold; the rate of attack then increases with increasing concentration of sulphate, but at a reducing rate of increase above about 1% concentration.
Also, the rate of attack will be faster if the sulphate is replenished, for example if the concrete is exposed to flowing groundwater. Concrete that has been attacked has a whitish appearance; damage usually starts at edges and corners, followed by progressive cracking and spalling, eventually leading to complete breakdown.
Although this stage can be reached in a few months in laboratory tests, it normally takes several years in the field. For any given concentration and type of sulphate, the rate and amount of deterioration decrease with:
- the C3A content of the cement, hence the low C3A content of sulphate-resisting Portland cement
- a higher cement content and lower water:cement ratio of the concrete; higher quality concrete is less vulnerable owing to its lower permeability. Figure 1 shows some results illustrating the combined effect of C3A content and concrete composition
- the incorporation of additions, which as we have seen can decrease the permeability and reduce the amount of free lime in the HCP, but also ‘dilute’ the C3A.
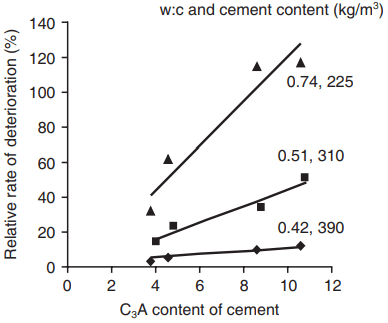
and concrete mix proportions on the deterioration in a soil containing 10% Na2SO4 (after Verbeck, 1968).
Recommendations for suitable concrete for use in sulphate-containing environments follow from a detailed knowledge of the above factors. For example, the relevant European specification, BS EN 206, classifies groundwater and soil as slightly, moderately or highly aggressive (denoted as exposure classes XA1, XA2 and XA3, respectively) according to the sulphate (SO42- ), carbon dioxide (CO2), ammonium (NH4+ ) and magnesium (Mg2+ ) contents and pH level, as shown in Table 1 for groundwater, thus encompassing sulphate and acid attack.
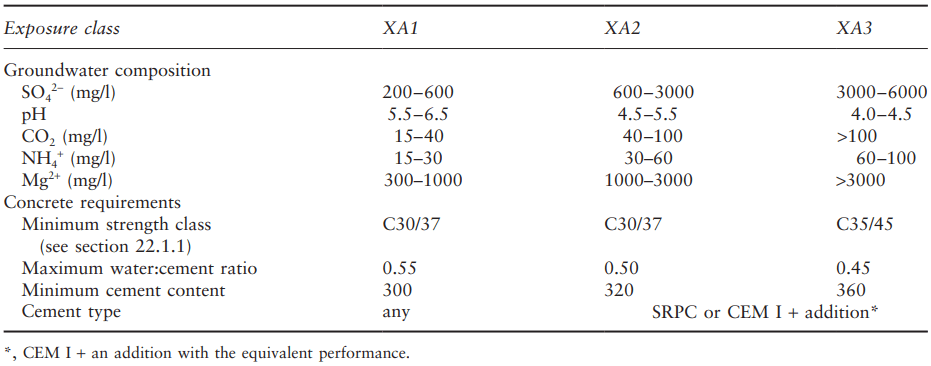
The concrete requirements show that lower sulphate levels do not require any special considerations over and above those for all concrete construction. With increasing sulphate levels combinations of sulphate resistance of the binder and overall quality of the concrete are required.
In the UK, the BRE (2005) have published more comprehensive guidelines for concrete in aggressive ground that include provision for the service requirements of the structure, the thickness of the concrete and for situations where the exposure conditions are so severe that concrete alone cannot be made sufficiently durable and extra measures, such as surface protection, are required.
These have been included in the relevant British Standard specification (BS 8500 part 1, 2006). We should also mention here delayed ettringite formation (DEF). The ettringite that is formed from the aluminates during cement hydration at normal temperatures breaks down at temperatures higher than about 70°C, and both the sulphates and aluminates appear to be reabsorbed by the C-S-H.
After cooling, the sulphate becomes available and ettringite is re-formed. By this time the cement paste has hardened, so the formation of expansive ettringite can lead to disruption. This is therefore a form of ‘internal sulphate attack’.
High temperatures may be deliberately applied to pre-cast concrete to increase strength gain, or more commonly may be the result of heat of hydration effects in large pours. The damage may not become apparent until some considerable time after casting.
DEF can be avoided by reducing the temperature rise, the use of fly ash or ggbs being particularly useful as their effect on the chemistry and microstructure of HCP also seems to be beneficial.
The Thaumasite Form of Sulphate Attack
This form of attack, known as TSA for short, also involves sulphates, but has distinct differences from the sulphate attack described above, and can have more serious consequences. Thaumasite is a rare mineral that occurs naturally in some basic rocks and limestones. It is a compound of calcium silicate, carbonate and sulphate with the formula CaSiO3.CaCO3.CaSO4.15H2O. To be formed in concrete and mortar it requires:
- a source of calcium silicate, clearly available from the hydrated or unhydrated Portland cement
- a source of sulphate ions, for example from soil or groundwater
- a source of carbonate, most commonly limestone aggregate or fillers or formed from the bicarbonates arising from the atmospheric carbon dioxide dissolved in pore water
- a wet, cold (below 15°C) environment.
Clearly all these requirements do not often occur together, the most common case where they do being in concrete made with limestone aggregate used for the foundations of structures in sulphate-bearing soils in temperate or cold climates.
Sulphides (e.g. pyrites, Fes2) can also be a problem, as when they are exposed to the atmosphere when the soil is disturbed during excavation they will oxidise to form more sulphates. Because the attack involves the calcium silicate hydrates, it can lead to complete disintegration of the cement paste, which turns into a soft, white, mushy mass.
The features of a concrete member that has been affected by TSA are a surface layer of the mushy mass, below which are regions that contain progressively decreasing amounts of thaumasite in cracks and voids, particularly around the aggregate particles. As well as reducing the strength of the concrete, the loss of effective cover to any reinforcing steel makes this more vulnerable to corrosion induced by, say, chloride penetration.
Incidents of attack are significant but not widespread, the most notable involving buried concrete in house and bridge foundations in the west of England, column building supports in Canada, tunnel linings, sewage pipes and road sub-bases. The rate of attack is generally slow and the incidents that came to light in England during the 1990s were in structures that were several years old.
These led to a major investigation, which included surveys of potentially vulnerable structures and laboratory studies. The reports of the investigation (Thaumasite Expert Group, 1999 and Clarke and BRE, 2002) make interesting reading, not only on the subject itself but also as an illustration of how government, industry and universities react together to a problem of this nature.
Following these extensive investigations, the most recent guidelines for concrete in aggressive ground published by the BRE (2005) do not include TSA as a separate consideration from other forms of sulphate attack. These differ from the previous recommendations because it has become apparent that the carbonates required for TSA can arise from atmospheric carbon dioxide (Collett et al., 2004), and hence protection against TSA is obtained by stricter requirements for all classes of exposure, rather than by treating it as a special case when limestone aggregates are used.
Seawater Attack
Concrete in seawater is exposed to a number of possible degradation processes simultaneously, including the chemical action of the sea salts, wetting and drying in the tidal zones and just above, abrasion from waves and water-borne sediment and, in some climates, freezing and thawing.
The total soluble salt content of seawater is typically about 3.5% by weight, the principal ionic contributors and their typical amounts being Cl– , 2.0%; Na+ , 1.1%; SO42- , 0.27%; Mg2+ , 0.12%; and Ca2+ , 0.05%.
The action of the sulphate is similar to that of pure sulphate solutions described above, with the addition of some interactive effects. Importantly, the severity of the attack is not as great as for a similar concentration of sulphate acting alone, and there is little accompanying expansion.
This is due to the presence of chloride ions; gypsum and ettringite are more soluble in a chloride solution than in pure water, and therefore tend to be leached out of concrete by seawater, and hence their formation does not result in expansive disruption.
Magnesium sulphate also participates in the reactions as in equation 5, and a feature of concrete damaged by seawater is the presence of white deposits of Mg(OH)2, often called brucite.
In experiments on concrete permanently saturated with seawater, a form of calcium carbonate called aragonite has also been found, arising from the reaction of dissolved carbon dioxide with calcium hydroxide. The brucite and aragonite can have a pore-blocking effect, effectively reducing the permeability of the concrete near the surface (Buenfeld and Newman, 1984).
In areas subject to wetting and drying cycles, salts will crystallise as the water evaporates, which can lead to a high salt concentration on or in the concrete’s surface and to potential disruption from the pressure exerted by the crystals as they rehydrate and grow during subsequent wetting–drying cycles – a process known as salt weathering.
This can be compounded by damage from freeze–thaw cycles or wave action, depending on the environment. These areas therefore tend to be the most vulnerable. The key to the elimination or at least reduction of all these problems is, not surprisingly, the use of a low-permeability concrete, perhaps combined with some limits on the C3A content of the cement, or the use of additions.
However, for the reasons given above, the degradation processes in many climates do not cause rapid deterioration, which explains why concrete of even relatively modest quality has a long and distinguished history of use in marine structures, both coastal and offshore. The salts in seawater can contribute to two other, potentially much more critical, degradation processes, namely alkali–aggregate reaction and corrosion of embedded steel.
Acid Attack
We have seen that the hardened cement paste binder in concrete is alkaline, and therefore no Portland cement concrete can be considered acid resistant.However, it is possible to produce a concrete that is adequately durable for many common circumstances by giving attention to low permeability and good curing.
In these circumstances, attack is only considered significant if the pH of the aggressive medium is less than about 6. Examples of acids that commonly come into contact with concrete are dilute solutions of carbon dioxide (CO2) and sulphur dioxide (SO2) in rain water in industrial regions, and carbon dioxide (CO2) and hydrogen sulphide (H2S)-bearing groundwater from moorlands.
The acids attack the calcium hydroxide within the cement paste, converting it, in the case of CO2, into calcium carbonate and bicarbonate. The latter is relatively soluble, and leaches out of the concrete, destabilising it.
The process is thus diffusion controlled, and progresses at a rate approximately proportional to the square root of time. The C-S-H may also be attacked, as can calcareous aggregates such as limestone. The rate of attack increases with reducing pH.
As mentioned above, the quality of the concrete is the most important factor in achieving acid resistance, but well-cured concretes containing additions also have greater resistance owing to the lower calcium hydroxide content as a result of the pozzolanic reaction.
In cases where some extra acid resistance is required, such as in floors of chemical factories, the surface can be treated with diluted water glass (sodium silicate), which reacts with the calcium hydroxide forming calcium silicates, blocking the pores.
In more aggressive conditions, the only option is to separate the acid and the concrete by, for example, applying a coating of epoxy resin or other suitable paint system to the concrete.
Alkali–Aggregate and Alkali–Silica Reaction
We described the general nature and composition of natural aggregates in former articales. Among many other constituents, they may contain silica, silicates and carbonates, which in certain mineral forms can react with the alkaline hydroxides in the pore water derived from the sodium and potassium oxides in the cement.
The general term for this is alkali– aggregate reaction (AAR), but the most common and important reaction involves active silica, and is known as the alkali–silica reaction (ASR). The product is a gel, which can destroy the bond between the aggregate and the HCP, and which absorbs water and swells to a sufficient extent to cause cracking and disruption of the concrete.
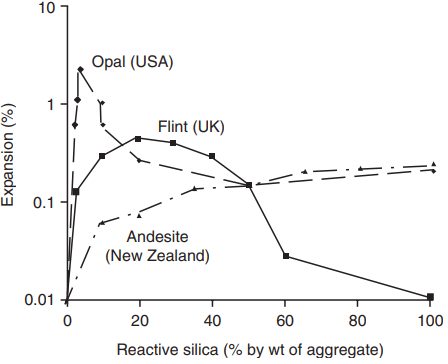
Compared to most other forms of degradation it is particularly insidious, as it starts within the concrete from reactions between the initial constituent materials. For the reaction to occur, clearly both active silica and alkalis must be present. In its reactive form, silica occurs as the minerals opal, chalcedony, crystobalite and tridymite and as volcanic glasses.
These can be found in some flints, limestones, cherts and tuffs. The sources of such aggregates include parts of the USA, Canada, South Africa, Scandinavia, Iceland, Australia, New Zealand and the midlands and southwest of England. Only a small proportion of reactive material in the aggregate (as low as 0.5%) may be necessary to cause disruption to the concrete.
In unhydrated cement, sodium and potassium oxides (Na2O and K2O) are present in small but significant quantities, either as soluble sulphates (Na2SO4 and K2SO4) or as a mixed salt (Na,K)SO4. There is also a small amount of free CaO, which is subsequently supplemented by Ca(OH)2 (portlandite) from the hydration reactions of C3S and C2S.
During hydration, these sulphates take part in a reaction with the aluminate phases in a similar way to gypsum, the product again being ettringite. With sodium, potassium and hydroxyl ions going into solution, the resulting pH of the pore water is 13 – 14, higher than that of saturated calcium hydroxide solution alone. Alkalis may also be contributed by some admixtures, fly ash and ggbs, and by external sources such as aggregate impurities, seawater or road de-icing salts.
The reactions between the reactive silica and the alkalis in the pore solution within the concrete to form the alkali–silicate gel occur first at the aggregate–cement paste interface.
The nature of the gel is complex, but it is clear that it is a mixture of sodium, potassium and calcium alkali–silicates. It is soft, but imbibes a large quantity of water, possibly by osmosis, and the sodium and potassium silicates swell considerably (the calcium silicates are nonswelling).
The hydraulic pressure that is developed leads to overall expansion of the concrete and can be sufficient to cause cracking of the aggregate particles, the HCP and the transition zone between the two. Continued availability of water causes enlargement and extension of the cracks, which eventually reach the outer surface of the concrete, forming either ‘pop-outs’ if the affected aggregate is close to the surface, or more extensive crazing, or ‘map cracking’, on the concrete surface, as illustrated in Fig. 2.
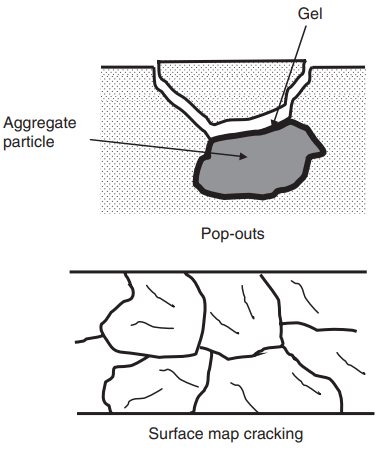
These surface cracks are often highlighted by staining from the soft gel oozing out of them. In general, cracking adversely affects the appearance and serviceability of a structure before reducing its load-carrying capacity.
The whole process is often very slow, and cracking can take years to develop in structural concrete. A description was first published in the USA by Stanton in 1940, since when numerous examples have been reported in many countries. Over 100 cases were identified in the UK between 1976 and 1987, triggering much research aimed at understanding and quantifying the mechanisms involved, determining its effects on structural performance and providing guidance for minimising the risk in new concrete.
The latter can be considered successful, as there have been very few, if any, confirmed cases of ASR in the UK since 1987. Laboratory tests on ASR often take the form of measuring the expansion of concrete or mortar specimens stored over water at 38°C to accelerate the reactions.
The mortar is made with crushed aggregates, thereby speeding up any reaction and expansion, but there are sometimes conflicting results from the two methods.
Even though such tests have not always satisfactorily explained all field observations, the most important factors influencing the amount and rate of reaction can be summarised as follows:
The amount of alkali available. Since sodium and potassium ions react in a similar way, it is normal practice to convert the amount of potassium oxide in a cement to an equivalent amount of sodium oxide (by the ratio of the molecular weights) and to express the alkali content as the weight of sodium oxide equivalent (Na2Oeq), calculated as:
Na2Oeq = Na2O + 0.658.K2O (6)
If the aggregates contain any salts, then their contribution can be obtained from the sodium oxide equivalent of the measured chloride content:
Na2Oeq = 0.76.Cl– (7)
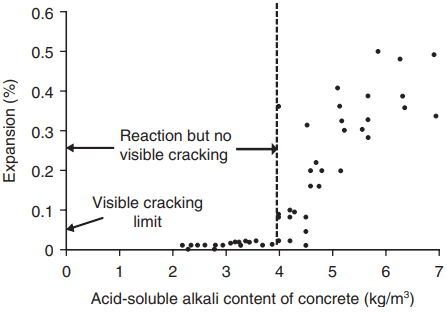
Stanton’s early work on ASR showed that expansion is unlikely to occur if the alkali content of the cement is less than about 0.6% Na2Oeq. Such cements are often called ‘low-alkali cements’.
expansion and cracking after 200 days in alkali–silica
reaction tests (after Hobbs, 1986).
More recent tests, which varied the alkali content of the concrete (Fig. 3) indicate that there is a threshold level (typically about 3.5 – 4 kg/m3 of concrete) below which no disruption will occur, even with reactive aggregates.
aggregates on the expansion of concrete from the
alkali–silica reaction
The amount of active silica. Some results from tests on concrete are shown in Fig. 4. The expansion increases with active silica content in the aggregate, but in two of the three sets of results only up to a certain content, beyond which the expansion reduces.
There is thus a pessimum ratio of silica:alkali for maximum expansion, which varies for different combinations of materials, but is probably the point at which the amount of reactive silica is just sufficient to react with all the alkali present. The ratio usually lies in the range 3.5 to 5.5.
One explanation is that at high silica contents, above the pessimum, greater proportions of the sodium and potassium are tied up, reducing the pH and increasing the amount of the non-expansive calcium alkali silicate produced.
The aggregate particle size, which affects the amount of reactive silica exposed to the alkali; fine particles (20 – 30 mm) can lead to expansion within a few weeks, larger ones only after many years.
The availability of moisture. Gel swelling will cease if the relative humidity within the concrete, which depends on the environment and the concrete’s permeability, falls below about 85%. Alternate wetting and drying may be the most harmful situation, possibly because it can lead to local high concentrations of the reacting materials.
The ambient temperature. Higher temperatures accelerate the reaction, at least up to 40°C.
The presence of additions, e.g. fly ash, ggbs or microsilica, and lithium salts. Microsilica is particularly effective (Fig. 5). Even though fly ash and ggbs themselves contain quantities of alkalis, if the rate of addition is sufficiently high then these do not contribute to the formation of the gel and so need not be taken into account.
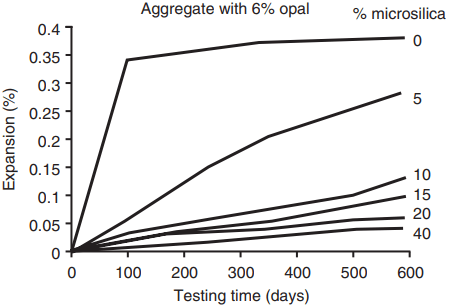
binder on the expansion of concrete from the alkali–
silica reaction
It seems that the hydrates formed in the secondary pozzolanic reaction bind the alkalis, either from the cement or contained in the additions, which are therefore not available to react with the aggregate.
However, the exact mechanisms and quantitative nature of the role of additions are complex and still unclear. Once started, the only effective way of stopping ASR is by eliminating water and ensuring that the concrete remains dry throughout its life; this is clearly impractical in many structural situations.
It follows that it is important to reduce or eliminate the risk of ASR occurring by careful selection of materials and concrete mix design. Consideration of the factors influencing the occurrence and rate of ASR described above leads to the following possibilities:
- Avoiding the use of reactive aggregates. This can be more difficult than it sounds, particularly with mixed mineral aggregates.
- Testing aggregates for their potential reactivity. There are a number of tests for aggregate reactivity, several involving accelerated expansion of mortar or concrete. A concrete prism test that was developed in the UK (BS 812-123) has been used by many organisations in the UK, and the results obtained show good correlation with the field performance of aggregates.
- Limiting the amount of alkali in the cement, for example by using a low-alkali cement, i.e. one with an alkali content of less than 0.6% by weight, as discussed above.
- Combining the Portland cement with an addition. However, to be wholly effective significant quantities must be added and minimum cement replacement levels of 25 – 40% fly ash, 40 – 50% ggbs and 8% microsilica have been suggested.
- Limiting the total alkali content of the concrete. Alkalis from all sources – cement, additions (but taking account of the factors discussed above), de-icing salts, etc. – should be safely below a threshold value such as that shown in Fig. 3.
BRE Digest 330 (Building Research Establishment, 2004) contains much more detailed explanations and comprehensive recommendations, and is a good example of the type of document that is of direct value to concrete practitioners.
Frost Attack – Freeze–Thaw Damage
In cold climates frost attack is a major cause of damage to concrete unless adequate precautions are taken. When free water in the larger pores within the HCP freezes it expands by about 9% and, if there is insufficient space within the concrete to accommodate this, then potentially disruptive internal pressures will result.
Successive cycles of freezing and thawing can cause progressive and cumulative damage, which takes the form of cracking and spalling, initially of the concrete surface. It is the water in the larger capillary pores and entrapped air voids that has the critical effect; the water in the much smaller gel pores is adsorbed onto the C-S-H surfaces, and does not freeze until the temperature falls to about -78°C.
However, after the capillary water has frozen it has a lower thermodynamic energy than the still-liquid gel water, which therefore tends to migrate to supplement the capillary water, thus increasing the disruption. The disruptive pressure is also enhanced by osmotic pressure.
The water in the pores is not pure, but is a solution of calcium hydroxide and other alkalis, and perhaps chlorides from road deicing salts or seawater; pure water separates out on freezing, leading to salt concentration gradients and osmotic pressures, which increase the diffusion of water to the freezing front.
The magnitude of the disruptive pressure depends on the capillary porosity, the degree of saturation of the concrete (dry concrete will clearly be unaffected) and the pressure relief provided by a nearby free surface or escape boundary. The extent of this pressure relief will depend on:
- the permeability of the material
- the rate at which ice is formed
- the distance from the point of ice formation to the escape boundary.
In saturated cement paste, the disruptive pressures will only be relieved if the point of ice formation is within about 0.1 mm of an escape boundary. A convenient way of achieving this is by the use of an air-entraining agent, which entrains air in the form of small discrete bubbles, and an average spacing of about 0.2 mm is required.
The capillary porosity of a cement paste or concrete, and hence its susceptibility to frost attack, can be reduced by lowering the water:cement ratio and ensuring that by proper curing hydration is as complete as possible. Bleeding, which results in local high-porosity zones, should also be minimised.
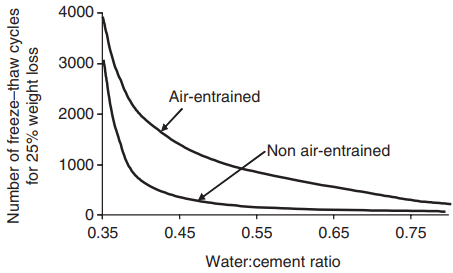
water:cement ratio on the freeze–thaw resistance of
concrete moist-cured for 28 days.
The combined effects of air entrainment and water:cement ratio are illustrated in Fig. 6. Certain aggregates are themselves susceptible to freeze–thaw damage, and their use must be avoided if a durable concrete is to be achieved. The first sign of damage caused by aggregate disruption is normally pop-outs on the concrete surface.
Vulnerable aggregates include some limestones and porous sand stones; these generally have high water absorption, but other rocks with high absorption are not vulnerable. Similar consideration of pore size and distribution as for cement paste apply to aggregates; for example, it has been found that pores of about 4 – 5 mm are critical, since these are large enough to permit water to enter but not large enough to allow dissipation of disruptive pressure.
Aggregate size is also a factor, with smaller particles causing less disruption, presumably because the average distance to an escape boundary on the aggregate surface is lower. The only satisfactory way of assessing an aggregate is by its performance when incorporated in concrete, using field experience or laboratory testing. Some of the recommendations for concrete exposed to freeze–thaw action in the UK (which has a relatively mild climate in this respect compared to many countries) are shown in Table 2.
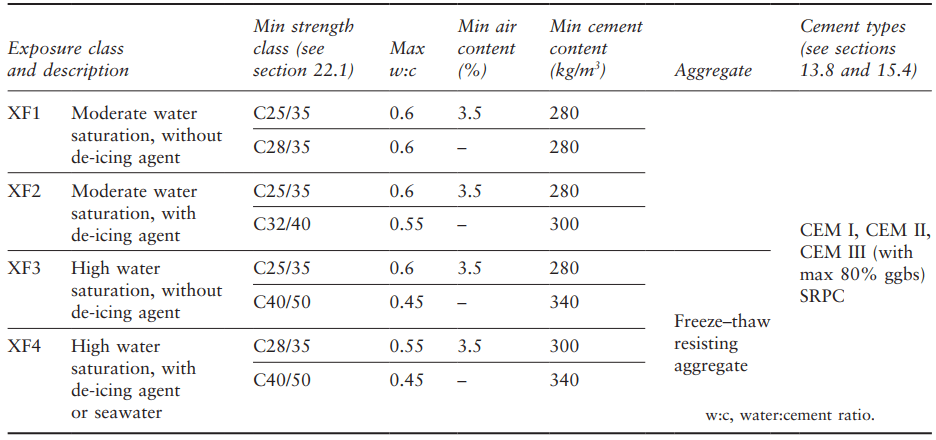
(from BS 8500, 2006).
For each exposure class this gives the option of air entrainment or a higher-quality concrete, and attention to the properties of the aggregate is required for the two most severe cases. The use of a de-icing agent, for example on exposed road surfaces, is included in the definition of the exposure class as it can result in distress from thermal shock when applied.
Fire Resistance
Concrete is incombustible and does not emit any toxic fumes when exposed to high temperatures. It is thus a favoured material, both in its own right and as protection for steelwork, when structural safety is being considered.
However, although it can retain some strength for a reasonable time at high temperatures, it will eventually degrade; the rate and amount of degradation depend on the maximum temperature reached, the period of exposure, the induced temperature gradients, the concrete’s constituents and moisture content and the size of the sample, and will therefore vary considerably.
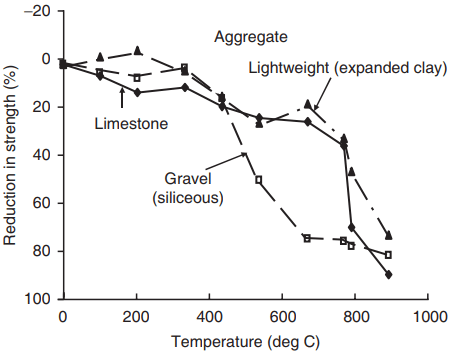
type on the compressive strength of concrete tested hot
(average initial strength, 28 MPa) (after Abrams, 1971).
Figure 7 shows typical results of testing small samples by holding them at elevated temperatures for a reasonable period of time. For temperatures up to about 500°C the reduction in strength is relatively gradual, but thereafter the decline is more rapid, giving almost total loss as the temperature approaches 1000°C. There are three main contributions to the degradation:
1. Evaporation of water within the concrete, which starts at 100°C, and continues with progressively more tightly held water being driven off. If the concrete is initially saturated and also of low permeability, then the water vapour cannot disperse quickly, and build-up of pressure can lead to cracking and spalling. This is therefore a particular problem with high-strength, low-porosity concrete.
Even though the total volume of water in the concrete is low, the induced pressures are very high, and progressive explosive spalling of the surface layers can occur within a few minutes of exposure to the fire. There have been some notable examples of damage of this type, most for example from two fires in heavy goods vehicles being carried on trains in the Channel Tunnel linking England and France, one in 1996 and one in 2008.
Both of these resulted in extensive damage to the high-strength concrete tunnel requiring costly and time-consuming repairs. The inclusion of polypropylene fibres in the concrete during mixing is one way of overcoming this effect; these rapidly melt and provide pressurerelief channels.
2. Differential expansion between the HCP and aggregate, resulting in thermal stresses and cracking, initiated in the transition zone. This is mainly responsible for the more rapid loss of strength above about 500°C, and also explains the superior performance of limestone and lightweight aggregate concrete apparent in Fig. 7; the former has a coefficient of thermal expansion closer to that of the HCP and the latter is less stiff and hence the thermal stresses are lower.
Lightweight aggregates have the additional advantage of decreasing the thermal conductivity of the concrete, thus delaying the temperature rise in the interior of a structural member.
3. Breakdown of the hydrates in the HCP, which is not complete until the temperature approaches 1000°C, but results in a total loss of strength at this point.
Thanks for reading about “degradation of concrete.”